
 moving within a cell.")
When Vamsi Mootha arrived at Harvard for medical school and found that New England weather was like nothing he’d known growing up in Texas, he was unhappy. Hoping to raise his spirits, his father’s cousin invited him to dinner at her apartment in Somerville. It was a snowy Friday evening, so he grabbed his thin coat and rode the T there, walking the last mile to her place, his sneakers becoming sodden as he trudged through the accumulating snow.
When he arrived, she gave him a towel to dry off. Then, as she made dinner, the storm became a blizzard, and the subway shut down. Mootha learned he would have to spend the night. After supper, he curled up with a textbook on a makeshift bed on the living room floor, and began to read—100 pages, and more the next day. “I ended up devouring that book.”
The subject was mitochondria, the tiny energy-producing organelles inside cells. Earlier that week, he’d read about them in his pathology class, for which the definitive textbook included just a single, brief reference suggesting that mutations in mitochondria might lead to a human muscle disease. “And for some strange reason, from that point onward, I just felt that this was my calling. Mitochondria were all I wanted to work on.”
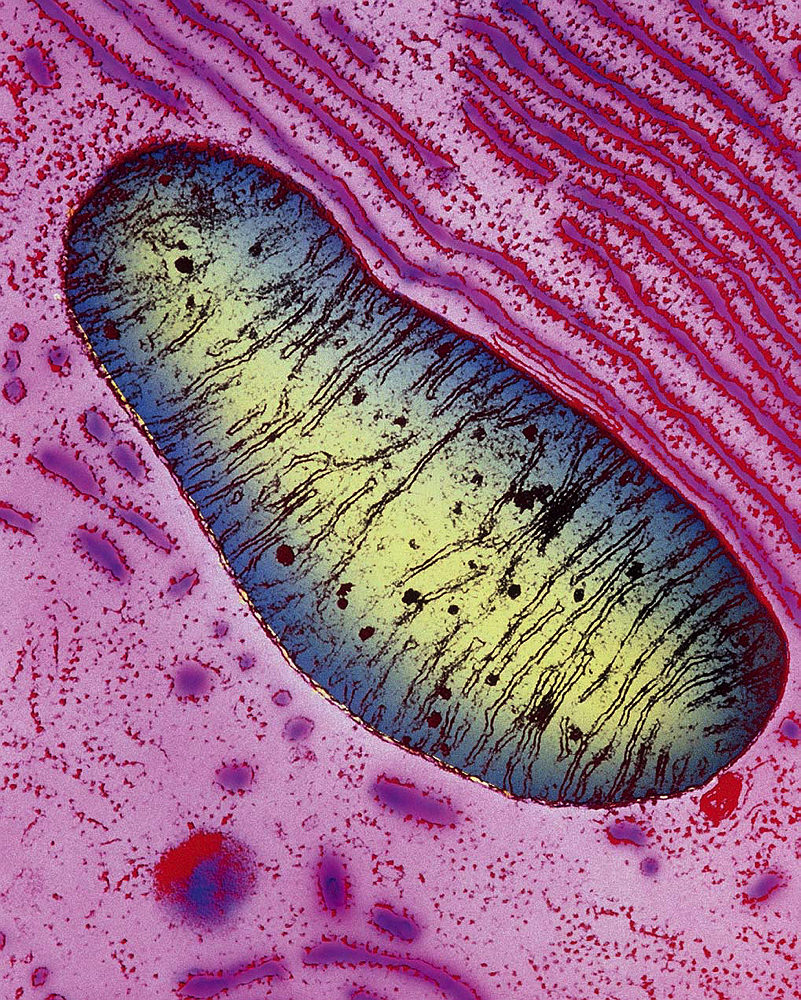
A mitochondrion within a pancreas cell. Mitochondria have enclosing outer membranes, and inner membranes with deep folds, called cristae, which extend far into the mitochondrial matrix. Chemical reactions that power cells occur on the cristae.
Image © Keith R. Porter/Science Source
A quarter-century later, Mootha, a former MacArthur Fellow, a Howard Hughes Medical Institute Investigator, and a professor of medicine and of systems biology at Harvard Medical School (HMS), runs a lab in the department of molecular biology at Massachusetts General Hospital dedicated “unapologetically” to the study of this organelle. Even in an era of specialization, such singular focus might seem extreme, but mitochondria are one of the most complex biological machines within cells—and arguably the most interesting. When functioning properly, they generate the fuel that cells burn. Mitochondrial dysfunction, on the other hand, is implicated in neurodegenerative disorders, diabetes, cancer, altered immune response, and even aging. But when asked why he studies these tiny intracellular machines, the first reason Mootha gives is aesthetic. “They’re beautiful,” he says, thumbing through a volume of scanning electron micrographs of the organelle. A few of these black and white images have been artificially tinted, revealing the varied shapes that mitochondria assume in different tissues. Colored red, blue and green, they’re jewel-like, with squiggles, waves, and zig-zags inside—the outlines of the inner membranes, called cristae.
Mitochondria produce metabolic energy by oxidizing carbohydrates, protein, and fatty acids. In a five-part respiratory chain, the organelle captures oxygen and combines it with glucose and fatty acids to create the complex organic chemical ATP (adenosine triphosphate), the fuel on which life runs. Cells can also produce a quick and easy form of sugar-based energy without the help of mitochondria, through an anaerobic process called glycolysis, but a mitochondrion oxidizing the same sugar yields 15 times as much energy for the cell to use. This energy advantage is generally accepted as the reason that, between one billion and one and a half billion years ago, a single free-living bacterium and a single-celled organism with a nucleus entered into a mutually beneficial relationship in which the bacterium took up residence inside the cell. No longer free-living, that bacterium evolved to become what is now the mitochondrion, an intracellular organelle.
Recent discoveries in the Mootha lab have suggested an alternative explanation for this unusual partnership that focuses on the organelle’s ability to detoxify oxygen by consuming it. But whatever the underlying reason, this ancient, extraordinary connection formed just once, Mootha says—and the evolutionary success it conferred was so great that the single cell multiplied and became the ancestor of all plants, animals, and fungi.
Nobody knows what that first cell looked like, but the bacterium that hitched a ride inside it was probably a relative of the “bugs” that cause Lyme disease, typhus, and chlamydia. In fact, mitochondria are similar enough to these bacteria that when physicians target such intracellular infections with specialized antibiotics, mitochondria are impaired, too. (These antibiotics, including the tetracyclines, are not dangerous for healthy people, Mootha says, but should be avoided by anyone with a mitochondrial disease.)
Feeling the Heat
The first documented case of mitochondrial disease involved a 30-year-old Swedish woman who arrived in May 1958 at the clinic of Rolf Luft, a physician at the Karolinska Institute near Stockholm, complaining that she constantly felt hot. The condition had begun when she was about seven; she had seen many doctors, but none had been able to pinpoint the cause. Luft measured elevated internal and skin temperature and noted that, although she ate constantly, she was thin and could not gain weight. To replace fluid lost through sweating, she had to drink large quantities of water. Although she was lethargic, her basal metabolic rate was double what is normal, and her heart was beating 100 times a minute. Luft biopsied her skeletal muscle and discovered that it was abnormally dense with mitochondria of large size and mass, packed with cristae.

Mitochondria in skunk cabbages generate enough heat to melt snow.
Photograph © Angelina Lax/Science Photo Library
But why was she hot? The process by which mitochondria create cellular energy, explains Mootha, releases heat. A famous example from the plant kingdom is skunk cabbage, which carries an especially inefficient form of mitochondria that can raise the plant’s temperature 30 degrees, melting snow and volatilizing the compounds it secretes to attract pollinators.
But there was nothing beneficial about the defect afflicting Luft’s patient. It had made her mitochondria nearly dysfunctional by channeling most of their energy to the production of heat. Doctors could offer her nothing but ice to keep her cool, and she eventually took her own life.
Researchers realized, however, that the ability to generate a moderate amount of heat could in some cases be advantageous. The thinking among scientists who study mitochondria, says Mootha, “is that there are very strong gene-environment interactions, so that certain types of mitochondrial variants may have been selected for in certain types of environments.” One theory holds that “perhaps inefficient genotypes may have been selected for in cold climates,” he notes, but rigorous evidence to support this claim is lacking right now. In a 2005 study using lab animals, however, researchers showed that variations in mitochondria (conferring long-distance running capability in one evolved population, and diabetes, obesity, and other metabolic ailments at the other extreme) can become fixed in as little as 11 generations, or 275 years in human terms, the blink of an eye on an evolutionary timescale. Although the Luft case predated the genomics era, researchers now understand that genetic mutations underlie mitochondrial diseases, which affect about 50,000 patients in the United States. As a class, these orphan diseases (pathologies too rare to attract market-driven pharmaceutical cures) are horrible for patients and vexing for physicians, because nobody knows how they work. “They’re super complex,” says Mootha, “because you can have two patients whose molecular defect is in the same part of the mitochondria,” yet one will be blind and deaf, and have neurodegeneration, disease of the heart muscle, and difficulty swallowing, while “the other may only have blindness, and every other organ system is fine.” This, he adds, “is one of the greatest mysteries, because all of our body’s tissues have mitochondria. They’re making energy in all of our body’s cells, but for some reason when you have defects in the mitochondrial proteome [the organelle’s full complement of proteins], sometimes many organ systems are impacted and then sometimes only one.”
Initially these diseases were called “maternally inherited syndromes” because mitochondrial DNA is inherited exclusively from mothers, all the way back to a common female human ancestor known as “mitochondrial Eve.” But as genetic understanding of the organelle improved, researchers realized that many mitochondrial diseases could be inherited from either parent, because most of the mitochondrial proteins are actually encoded by DNA in the cell nucleus, rather than by mitochondrial DNA. Furthermore, research has led to a growing realization that mitochondrial dysfunction is connected to a vast number of common diseases—including diabetes, heart disease, Parkinson’s and Alzheimer’s, hearing loss, and psychiatric disorders, including depression—affecting tens of millions of patients in the United States alone.
“The Coolest Machine of All”
Mootha has played a key role in this broadened understanding of mitochondria’s importance—and in the realization that the organelle’s role in energy production may be a red herring.
Trained at Stanford in mathematics and computer science, he studied mitochondrial biology for his thesis in the Harvard-MIT Health Sciences and Technology program (which integrates science, engineering, and medicine to solve problems in human health). Because there was “no lab at Harvard squarely focused” on the subject at the time, much of his graduate research took place at the National Institutes of Health in Maryland. He received his M.D. from HMS in 1998. During his subsequent clinical training at Brigham and Women’s Hospital (completed in 2001), his electives focused on patients with rare mitochondrial diseases.
Even then, Mootha knew that genes from mitochondrial DNA (mtDNA) alone could not account for the full range of mitochondrial diseases. Genes, he explains, encode the instructions for building proteins, which are the principal functional actors within cells—they get jobs done. The sequencing of the mitochondrial genome in 1981 (two decades before that of the human genome) had revealed that its mtDNA codes for just 13 proteins, nowhere near enough to explain the bulk of mitochondrial diseases. And that created a new mystery. Mitochondria, researchers knew, produce more than 1,000 proteins. Where was the DNA that encodes the bulk of those proteins?
The answer lies in evolutionary history. After mitochondria took up residence in cells more than a billion years ago—in what was probably a long, drawn-out process—many genes were transferred from the mitochondria into the genome of the host—in other words, into the nucleus of the cell, where most DNA resides. The result of this gene transfer is that the mitochondrial genome, with just 16,000 base pairs (the building blocks of DNA), has been stripped down to bare essentials. Compared to its ancestral form and also to living relatives, such as the Rickettsia bacterium that causes typhus, which has more than a million base pairs, its genome is now tiny.
In 2001, Mootha began his post-M.D. fellowship at the Whitehead Institute with Eric Lander, one of the leaders of the international project to sequence the human genome, officially completed in 2003. With a full sequence of the genes found in human-cell nuclei in hand, researchers anticipated being able, finally, to identify those genes that make the bulk of the proteins in mitochondria.
Now incorporated in each cell’s nuclear DNA, those ancient genes produce about two-thirds of all mitochondrial proteins. The other third, says Mootha, are the result of “innovations” that evolved after the initial union of bacterium and cell, and that now enable human mitochondria to “do things that no bacteria can do.”
Mootha has played a defining role in identifying all the mitochondrial proteins, both ancient and evolved, using a multidisciplinary approach that combines computational biology with biochemistry and the genetics of disease. “When I was completing my postdoc and interviewing for faculty positions, I knew that my work wouldn’t fit neatly into any traditional department,” he recalls. Fortunately, Marc Kirschner, now Enders University Professor of systems biology, had just founded a new HMS systems-biology department (see “From Physiology to Systems Biology”). Mootha was the first faculty member hired. “My goal was to achieve a holistic understanding of mitochondria…” by combining “the new tools of genomics with classical biochemistry,” he says. Systems biology, he explains, aims to “simultaneously understand how the machines of life evolve, develop, function, and fail. These are not independent goals—they must be tackled in parallel to be successful,” with a focus, in his case, “on the coolest machine of all.”
One of the many fundamental contributions of his lab has been to map all 1,158 of the mammalian mitochondrial genes that encode proteins. This proteome inventory (the entire set of expressed proteins in a organelle, cell, tissue, or organism), called the MitoCarta, was first published in 2008 and updated in 2015; it is free, readily available, and widely used by researchers worldwide. Mootha has used it to identify a major means by which mitochondria and their host cells communicate: via calcium signaling. His team has also used it to pinpoint more than a dozen genes linked to metabolic diseases, holding out the hope that these might be cured using gene therapy.* And he saw a pattern emerge that made him question whether the organelle’s role as a power plant could have blinded science to its other natural talent: devouring oxygen.
* The potential for gene therapy would at this time apply only to mutations in nuclear genes, Mootha notes; scientists don’t yet know how to perform gene therapy in mitochondrial DNA.
Toxic Oxygen?
Mootha began to wonder if energy production might not be the key to understanding the basis of mitochondrial disease. “When we look at these disorders, they’re not easily explained on the basis of energy requirements.” The organ systems most affected “are not necessarily the ones that have the highest energy demands.” An outpouring of research began to hint at the versatile, indispensable role that mitochondria play in the regulation of cell death, the immune system, and cell signaling. The traditional focus on energy production, in other words, may have misled researchers—all the way back to their interpretation of what happened more than a billion years ago, when that single cell and a lone bacterium entered into a long-term relationship.
Oxygen levels on early Earth were low at that time, but rising, Mootha says. “We think of oxygen as a life-giving molecule, and it is, but it can also be very corrosive”—think of how it rusts a car. In biology, oxygen and its byproducts are known to cause cellular damage, and are implicated in aging.
Mitochondria, on the other hand, are consumers of oxygen. Maybe, according to a hypothesis favored by the Mootha lab, the selective advantage that accrued to the first cell to host a mitochondrion was not only more energy, but better control of the toxic effects of oxygen. Normal gene expression supports this idea: “Our genes that turn on mitochondria,” says Mootha, “are really, really smart.” When turning on mitochondria, “the genes also turn on antioxidant programs.” And they calibrate antioxidant levels to the quantity of mitochondria that have been activated. “If you’re going to build a car that’s not an inline six but a V8 engine,” he points out, “you’re also going to need a bigger catalytic converter.”
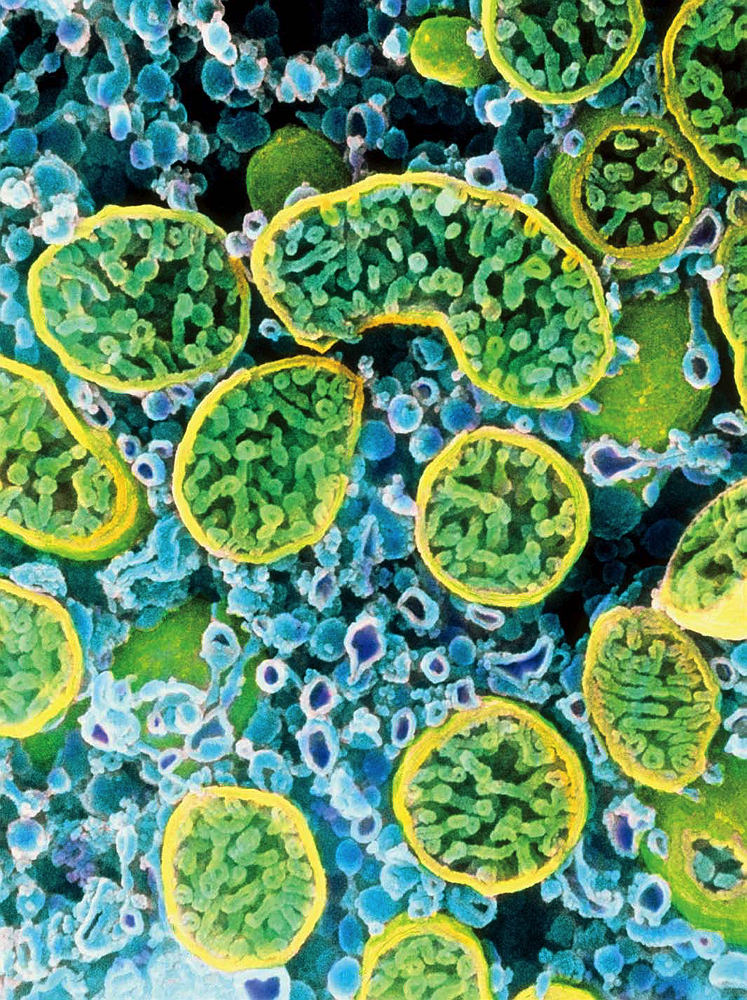
Cells actively secreting hormones, like the interior of this one, from an ovary, are dense with mitochondria (outlined in yellow).
Image © Professors P.M. Motta, S. Makabe and T. Naguro/Science Source
Interestingly, a 2009 study showed that the widespread practice of antioxidant vitamin supplementation can actually interfere with this natural response. For the experiment, researchers divided human subjects into four groups: exercisers who took antioxidant vitamins; exercisers who did not; non-exercisers who took antioxidant vitamins; and non-exercisers who did not. Both groups of exercisers were healthier after a few months. But surprisingly, those who exercised without the antioxidant vitamins did best, probably, Mootha speculates, “because the rest of the cell, when it senses some of these sparks, adapts in a way that is beneficial to the organism”—and does so better than with vitamins, which appeared to interfere with the health-promoting stress of physical exercise.
This result didn’t surprise him, because of the numerous adaptive responses to stress already known to be mediated through the mitochondria, evolved during a billion-year history. In this long view, any mutations that did not kill an individual cell (or the larger organism) might have opened the way for subsequent mutations that would bypass—or “rescue”—the damaged link in the chain of chemical reactions mitochondria use to make energy. In some cases, in other words, the evolved response of the organelle and host cell to the overloaded or damaged pathway can actually provide a net benefit to both the cell and the entire organism.
An interesting example of this overcompensation, Mootha says, occurs when diabetes patients are given Metformin, which interferes with normal mitochondrial function. The drug “has only one known target,” he explains: the first stage in the five-step process by which mitochondria produce energy. When that initial step is severely impaired, as in cases of mitochondrial disease, the results are devastating. But the weak inhibition caused by Metformin triggers an adaptive response that actually helps diabetes patients. “It’s a little bit like Mithridates, the Persian king who was afraid of getting poisoned, so he had his pharmacist mix all the poisons available and then took sublethal doses,” Mootha points out—and perhaps not all that different from a vaccination. Metformin induces “a state called hormesis, a protective response that’s net protective.” The effect is so promising that researchers recently began testing Metformin in human clinical trials to see whether hormesis can slow aging. The mechanism has been worked out at the genetic level in worms, but in higher organisms, he continues, “we actually don’t know what that program is right now. The mitochondrion is this beautiful organelle, but there are all these homeostatic and feedback loops within it” which are in turn “wired within the broader organism,” which has its own loops and feedback mechanisms.
With his computational background, Mootha has the tools and training to tease out the nature of this complexity, and use powerful, scaled approaches to develop new therapies. Inspired by the example of Metformin, he and colleagues initiated a genome-wide screen in 2014, with support from the Marriott Foundation. Searching for factors that, when disrupted, allow cells to cope with broken mitochondria, they got a hit. Their screen suggested that low levels of atmospheric oxygen could trigger a response that protects against Leigh syndrome, an inherited disease of the central nervous system—caused by mutations in any one of 75 different genes—that in children ends in death between the ages of three and 16 months, frequently as a result of respiratory failure.
When the researchers tested the idea in mouse models of mitochondrial disease, the results were astounding. While a normal mouse lives about two years, their diseased mice typically survived for just 55 days. But when the team lowered the oxygen concentration to 11 percent, a level typically found at altitudes of around 14,000 feet, they found that they could prevent the onset of the disease. The mice raised in those hypoxic conditions lived a full year. Even diseased mice on the brink of death could be revived by restricting oxygen. (They called that “the Lazarus effect.”). Extra oxygen, on the other hand, functioned like a poison, killing the mice within a few days.
Low-oxygen environments appear to confer benefits on humans, too. In 1975, Mootha points out, the Indian army reported distinct health effects on troops serving at 12,000 to 18,000 feet, along the Indochina border, as compared to those serving on the plains. In the short term, deaths from acute infections were much higher among the men posted at altitude, but over many years, “The incidence of new cases of diabetes, stroke, heart disease, and cognitive defects were dramatically reduced” compared to those who’d served at lower elevations. (Mootha adds the caveat that these results were purely observational, and that temperature, diet, and activity levels differed, too.) Epidemiological studies often rank Colorado, with a mean elevation of 6,800 feet, as the healthiest U.S. state.
The human data, combined with the rigorous mouse studies, he explains, suggest that “too much oxygen can actually be a bad thing.” Their mouse observations were soon buttressed by further anecdotes involving humans. After the team published the paper in 2016, he received calls from doctors in California and elsewhere who had been treating their patients with mitochondrial disease in hyperbaric chambers, giving them extra oxygen in an attempt to alleviate their symptoms. “Two young patients became comatose after this treatment, and never woke up,” he relates. Two others, who were legally blind in one eye but not the other, became blind in both eyes after hyperbaric oxygen exposure. Could it be, he asks, that “because the mitochondria aren’t consuming oxygen properly, there’s an excess that is corrosive to the rest of the cell?” This idea from Mootha’s lab may have broad application to health, particularly in diseases associated with aging, which is itself linked to mitochondrial decline.
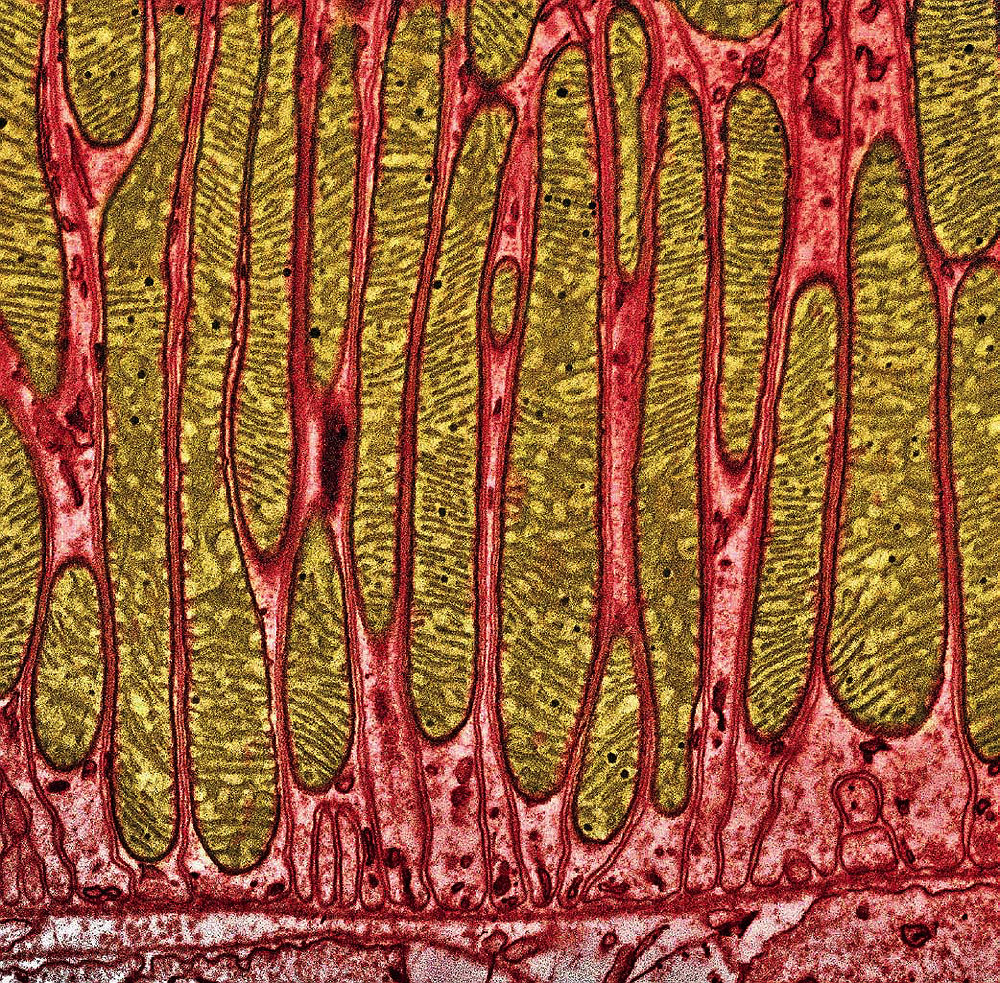
Elongated mitochondria (brown) are packed tightly in a kidney cell.
Image © Thomas Deerinck, NCMIR/Science Source
In humans, a typical cell might contain a few dozen mitochondria, joined together in a mass and working as one network. Liver and muscle cells, with their key roles in producing, storing, and using energy, can contain thousands of them. Regardless of the baseline, “The quantity of mitochondria declines as a function of age,” Mootha explains, which may increase people’s vulnerability to conditions ranging from Parkinson’s disease (in which mitochondrial dysfunction is causal) to diabetes. Furthermore, there is a double insult in older persons: their reduced numbers of mitochondria are less efficient than a young person’s. But exercise, at any age, can boost mitochondrial counts. “And when your skeletal muscle increases the number of mitochondria,” Mootha adds, “it actually eliminates some of the bad ones,” increasing overall efficiency, too. In fact, he says, “many of the beneficial effects of exercise and healthy diets are likely being mediated through the mitochondria.”
The larger context for Mootha’s career-long focus on a single organelle is his aspiration to develop a foundation for precise mitochondrial medicine. “There are three ingredients,” he says. “First, from a single tube of blood, we want to establish a molecular diagnosis,” using genetics. “Second, from that same vial, by analyzing the products of metabolism circulating in the blood (see “Fathoming Metabolism,” May-June 2011, page 27), we want to be able to gauge the severity of mitochondrial dysfunction. I’d love to be able to draw your blood and say, ‘I think you can do better, why don’t you exercise a bit more, why don’t you work on your diet,’ or even, when administering an experimental therapy targeting your mitochondria, I’d like to be able to know whether it is working.” Third, he wants to develop therapies, not only for rare, fatal mitochondrial disorders caused by spontaneous or inherited mutations, but for the range of common diseases in which mitochondrial dysfunction has recently been implicated. Identifying drugs that limit uptake or delivery of oxygen is an obvious next step.
He believes that “we’re just now scratching the surface of a very deep relationship between mitochondria and oxygen that has evolutionary origins.” If the logic of that relationship from a systems perspective can be understood, “then perhaps we can exploit it for therapeutic purposes”—either as a gas or a pill. “Hypoxia medicine,” he says, “may be our first foray” into therapeutics.
Having explained his research plan, Mootha rises to go. It is Patriots Day, April 16, in New England, and the weather is unseasonably cold, intensified by wind and driving rain. Mootha is heading out to the finish line of the Boston Marathon to cheer the father of a young girl with Leigh’s syndrome, running to support his lab. He is in high spirits, dressed head to toe in sensible raingear, seemingly unperturbed by the prospect of standing in the cold to wait. In the quarter-century since he first came to Boston, he has clearly adapted. Perhaps his mitochondria have, too.








